Carisma Therapeutics Announces Changes to its Board of Directors
Carisma Therapeutics Announces Changes to its Board of Directors
Appointment of John Hohneker, M.D.
Resignation of Chidozie Ugwumba
PHILADELPHIA, April 1, 2024 /PRNewswire/ — Carisma Therapeutics Inc. (Nasdaq: CARM) (“Carisma” or the “Company”), a clinical-stage biopharmaceutical company focused on discovering and developing innovative immunotherapies, today announced the appointment of John Hohneker, M.D. to the Board of Directors of the Company, effective April 1, 2024. Additionally, the Company announced that Chidozie Ugwumba, who has served on Carisma’s Board of Directors since December 2020, has advised the Board of his intent to step down from his role as a member of the Board and chair of the Audit Committee of the Board effective April 1, 2024, as a result of his other professional commitments.
“We are pleased to welcome John to the Carisma Board of Directors,” said Sanford Zweifach, Chair of the Carisma Board of Directors. “I believe John’s extensive track record of clinical development, strategic leadership, and public company Board membership in the biopharmaceutical space will enable him to immediately support the Company as it continues leading the field of engineered macrophage development. We would also like to thank Chidozie for his invaluable advice and guidance since he first joined the Board over three years ago.”
“I am excited to join Carisma’s Board as the Company continues to develop innovative immunotherapies,” said Dr. Hohneker. “I am looking forward to partnering with the Board and the management team as they work to potentially bring novel treatments to patients in need across multiple therapeutic areas.”
Dr. Hohneker brings over 30 years of extensive experience in drug development and leadership across the biotech and pharmaceutical sectors. He most recently served as President and Chief Executive Officer of Anokion SA. Prior to this, he held the role of President of Research and Development at FORMA Therapeutics Inc. Before joining FORMA Therapeutics, Dr. Hohneker held roles of increasing responsibility at Novartis AG, including most recently as Senior Vice President and Global Head of Development, Immunology, and Dermatology. Prior to his time at Novartis, Dr. Hohneker held positions of increasing responsibility at Glaxo Wellcome and its legacy company, Burroughs Wellcome.
Dr. Hohneker currently serves on the Board of Directors of Curis, Inc., Sonata Therapeutics, Inc., and Trishula Therapeutics, Inc. Previously, Dr. Hohneker has served as a member of the Board of Directors of Torque Therapeutics, Inc., Dimension Therapeutics, Inc., Cygnal Therapeutics, Inc., BioTheryX Inc., Evelo Biosciences, Inc., Humanigen, Inc. and Aravive, Inc. Dr. Hohneker received his bachelor’s degree in chemistry from Gettysburg College and his M.D. from the Rutgers School of Biomedical and Health Sciences (formerly the University of Medicine and Dentistry of New Jersey-Rutgers Medical School). He completed his internship and residency in internal medicine and his fellowship in medical oncology, all at the University of North Carolina at Chapel Hill.
About Carisma
Carisma Therapeutics Inc. is a clinical-stage biopharmaceutical company focused on utilizing our proprietary macrophage and monocyte cell engineering platform to develop transformative immunotherapies to treat cancer and other serious diseases. We have created a comprehensive, differentiated proprietary cell therapy platform focused on engineered macrophages and monocytes, cells that play a crucial role in both the innate and adaptive immune response. Carisma is headquartered in Philadelphia, PA. For more information, please visit www.carismatx.com.

TRiCares Announces the Appointment of Ahmed Elmouelhi as President & Chief Executive Officer
Paris, France and Munich, Germany, March 25, 2024 – TRiCares SAS (“TRiCares”) a privately held pioneer in the field of minimally invasive treatment of tricuspid regurgitation, today is pleased to announce the appointment of Ahmed Elmouelhi as President & Chief Executive Officer (CEO). In conjunction with this, and following eight years at the helm of the business, Helmut Straubinger is retiring as President & CEO.
Ahmed Elmouelhi is a seasoned professional with more than 20 years of medical device experience across strategy, sales and marketing, R&D, and clinical science. Before joining TRiCares, Ahmed served as the Senior Vice President of Product Marketing, Strategy, and Business Development at AtriCure. There, he was instrumental in the establishment of Left Atrial Appendage (LAA) therapies and the creation of two successful business franchises for the company. Previously, Ahmed held roles of increasing responsibility at Medtronic, within its Structural Heart division, as well as AGA Medical (now part of Abbott Laboratories), where he led cross-functional teams to launch multiple generations of Transcatheter Aortic Valve Replacement (TAVR) therapies and accessories, Congenital Heart Defect therapies, and percutaneous LAA devices.
Ahmed studied at Massachusetts Institute of Technology where he obtained both his Masters and Bachelors in Mechanical Engineering with a minor in Economics. He currently serves as the Chairman of the American Heart Association in Minnesota.
Since joining the business in March 2016, Helmut has overseen the development of the first European minimally invasive implantable tricuspid valve, bringing it to first-in-man clinical use. Helmut will work with Ahmed to provide a seamless transition before his retirement at the end of March and will continue to support the team on a consultancy basis.
Eric Fain, MD, Chairman of TRiCares, commented: “I would like to extend my thanks to Helmut, on behalf of the board of directors, for his efforts over many years in leading TRiCares to successfully develop the leading transcatheter tricuspid replacement technology and demonstrate its initial strong clinical performance. After evaluating many candidates for the next CEO, I am confident that Ahmed is the right person to drive the company’s success as it enters its next phase.”
Ahmed Elmouelhi, newly appointed President & CEO of TRiCares, added: “This is an exciting time to be joining the business, and I am looking forward to leading the team over the coming months and years. The potential for Topaz is clear, and the need great, with 1.8 million patients suffering from Tricuspid heart valve regurgitation in the US alone. Helmut leaves a significant legacy after many years with the business, and I wish him the best for his retirement.”
Helmut Straubinger, former President & CEO of TRiCares, commented: “I would like to express my heartfelt gratitude to the entire TRiCares team for their unwavering commitment and hard work over the last eight years. As the company looks towards the US, this is the right moment for a change in leadership. I look forward to following TRiCares’ progress under Ahmed’s guidance, as it seeks to benefit Tricuspid regurgitation patients globally.”
Press inquiries:

Bayer and Aignostics to collaborate on next generation precision oncology
Bayer and Aignostics to collaborate on next generation precision oncology
- Strategic multi-year research collaboration to identify novel targets with strong disease links and to accelerate clinical development, further strengthening Bayer’s precision oncology development portfolio
- Co-development of a novel target identification platform leveraging multimodal patient data and industry-leading AI/ML algorithms
- Development of computational pathology algorithms connecting baseline data such as molecular tumor profiles with patient outcome to enable better patient identification, stratification, and selection for clinical trials
Berlin, Germany, March 14, 2024 – Bayer and Aignostics GmbH today announced a strategic collaboration on several artificial intelligence (AI)-powered approaches with applications in precision oncology drug research and development. Aignostics is a spin-off from one of the world’s leading hospitals, Charité-Universitätsmedizin Berlin, and a global leader in using computational pathology to transform complex biomedical data into biology insights.
The partners will co-create a novel target identification platform that leverages Aignostics’ technology and proprietary multimodal patient cohorts, and Bayer’s deep expertise in discovering and developing novel oncology therapies. In addition, the collaboration will include the development of computational pathology algorithms powered by AI and machine learning (ML) that connect baseline pathology data, such as molecular tumor profiles, with clinical data, such as patient outcomes, to enable better patient identification, stratification, and selection for clinical trials.
The goal of the multi-year research collaboration is to identify novel cancer targets with a strong disease link through AI models applied to multimodal patient data and to accelerate clinical development of oncology programs. This approach has the potential to address some of the challenges currently experienced in target discovery and disease heterogeneity.
“Gaining insights into human disease biology, discovering targets with a strong disease link by integrating artificial intelligence, machine learning and multimodal pathology into precision drug development has a huge potential for our R&D innovation strategy,” said Christian Rommel, Member of the Executive Committee of Bayer’s Pharmaceuticals Division and Head of Research and Development. “Bringing Aignostics’ technical knowhow and their access to large patient datasets together with Bayer’s expertise in cancer research and development will enable discoveries and faster clinical development, helping to provide cancer patients with more impactful medicines.”
The collaboration will leverage Aignostics’ technology and access to longitudinal, multimodal clinical data sets in well characterized patient cohorts to discover new oncology targets for high unmet medical need indications. Under the terms of the agreement, the companies will collaborate on multiple discovery programs and initiate at least two target identification programs.
“Innovation at Aignostics has always been fueled by close collaboration with clinicians and biopharma. With this partnership, we’re thrilled to take that approach to the next level. Fusing our technology and multimodal data with Bayer’s extensive expertise in drug discovery and clinical development has the potential to generate better drugs for patients with high unmet need in less time. Together with Bayer, we’re excited to transform AI’s immense potential into a reality for healthcare,” said Viktor Matyas, CEO of Aignostics GmbH.
Aignostics will receive an upfront payment and is eligible to receive success-based milestone payments and royalties on any commercialized therapies that result from the collaboration.
About Aignostics
Aignostics combines proprietary access to multimodal clinical datasets, industry-leading AI technologies, and rigorous science to develop best-in-class insights for the next generation of precision medicine. Through collaborations with its biopharma partners, Aignostics supports drug discovery, translational research, clinical trials, and CDx development across multiple therapeutic areas. Established in 2018, Aignostics is a spin-off from Charité Berlin, one of the world’s largest and most esteemed university hospitals. Aignostics is funded by leading VC investors and has operations in Berlin and New York.
About Bayer
Bayer is a global enterprise with core competencies in the life science fields of health care and nutrition. In line with its mission, “Health for all, Hunger for none,” the company’s products and services are designed to help people and the planet thrive by supporting efforts to master the major challenges presented by a growing and aging global population. Bayer is committed to driving sustainable development and generating a positive impact with its businesses. At the same time, the Group aims to increase its earning power and create value through innovation and growth. The Bayer brand stands for trust, reliability and quality throughout the world. In fiscal 2023, the Group employed around 100,000 people and had sales of 47.6 billion euros. R&D expenses before special items amounted to 5.8 billion euros. For more information, go to www.bayer.com.
Bayer Media Contact:
Julia Schulze, phone +49 175 5866 432
Email:
Aignostics Media Contact:
MC Services AG, Kaja Skorka, Dr. Regina Lutz, phone: +49 89-210 2280
Email:
Find more information at https://pharma.bayer.com/
Follow us on Facebook: http://www.facebook.com/bayer
Follow us on Twitter: @BayerPharma
Find more information at https://aignostics.com/
Follow us on LinkedIn: https://www.linkedin.com/company/aignostics
jds (2024-0053E)
Forward-Looking Statements
This release may contain forward-looking statements based on current assumptions and forecasts made by Bayer management. Various known and unknown risks, uncertainties and other factors could lead to material differences between the actual future results, financial situation, development or performance of the company and the estimates given here. These factors include those discussed in Bayer’s public reports which are available on the Bayer website at www.bayer.com. The company assumes no liability whatsoever to update these forward-looking statements or to conform them to future events or developments.
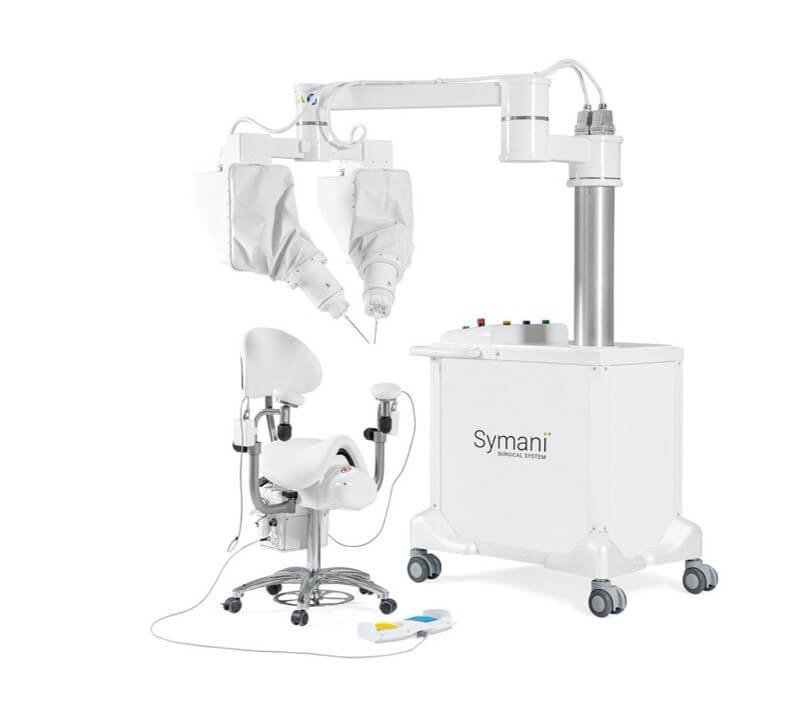
MMI Raises $110 Million in Series C Financing
MMI Raises $110 Million in Series C Financing
Largest ever investment in microsurgery will further MMI’s mission to transform open surgery with robotic technology
JACKSONVILLE, Fla.–(BUSINESS WIRE)–MMI (Medical Microinstruments, Inc.), a robotics company dedicated to increasing treatment options and improving clinical outcomes for patients with complex conditions, today announced that it has raised $110 million in Series C financing. The round, led by Fidelity Management & Research Company, marks the largest ever investment in microsurgery innovation.
The funds will support commercialization of the Symani® Surgical System in high-growth markets and continued investment in studies that generate clinical evidence and enable indication expansion. Investments will also accelerate advanced technology capabilities and enable MMI to scale its operational capabilities globally.
MMI and its existing investors, all of whom contributed to the Series C financing, see considerable opportunity for rapid growth. The company projects the market for eligible robotic microsurgical procedures will grow from 3 million to 22 million annually by 2028, driven primarily by technological advancements and indication expansion.
“Against a backdrop of plateauing investments in medical robotics, this support builds on our confidence in a new, less invasive solution for open surgery, a significant market that can benefit from the smallest wristed microinstruments,” said Mark Toland, CEO of MMI. “Our Symani Surgical System is uniquely positioned to expand patient access to care by accelerating the number of surgeons able to perform complex, delicate procedures. With the support of our investors, we will continue to advance our technology through a growing body of clinical evidence and expanded hospital partnerships.”
The Symani Surgical System is a first-of-its-kind robotic technology that uniquely addresses the scale and complexities of microsurgery and supermicrosurgery. By allowing surgeons to replicate the natural movements of the human hand at the micro scale, it can expand treatment options for patients in need of soft tissue, open surgical procedures, such as free flap reconstructions, lymphatic surgery, and trauma replantations. It is designed to help restore quality of life for more patients, accelerate the number of surgeons able to push the boundaries of complex procedures for delicate anatomy, and enable hospitals to expand their open surgical programs.
MMI has raised over $200 million in funding to date. In 2022, it closed a Series B financing round to propel growth. The funding was allocated to help expand indications and support ongoing commercialization efforts for the Symani Surgical System in Europe, where it received CE mark in 2019, accelerate plans to commercialize in the U.S. and Asia-Pacific, and advance clinical research.
To learn more about MMI and the Symani Surgical System, visit MMI’s website here: https://mmimicro.com.
About MMI
MMI (Medical Microinstruments, Inc.) is on a mission to advance robotic technology that pushes the limits of soft tissue open surgery and opens new opportunities for surgeons to restore quality of life for more patients with complex conditions. The company was founded in 2015 near Pisa, Italy, and its proprietary Symani® Surgical System combines the world’s smallest wristed microinstruments with tremor-reducing and motion-scaling technologies to address significant unmet patient needs across the globe. This first-of-its-kind surgical robotic platform for open, soft tissue micro-level surgery can help address microvascular repair, lymphatic repair, and peripheral nerve repair. The Symani System is CE Marked for commercial use in Europe. In the United States, the system is not approved or cleared for commercial use. MMI is backed by global investors including Fidelity Management & Research Company, Andera Partners, BioStar, Deerfield Management, Fountain Healthcare Partners, Panakès Partners, RA Capital, Sambatech, and Wellington Partners.


UroMems Announces Results of First-Ever Smart Artificial Urinary Sphincter Implant in Female Patient to Treat Stress Urinary Incontinence
UroMems Announces Results of First-Ever Smart Artificial Urinary Sphincter Implant in Female Patient to Treat Stress Urinary Incontinence
Positive results demonstrate promising new option for women seeking better, personalized treatment
GRENOBLE, France and MINNEAPOLIS, Feb. 14, 2024 /PRNewswire/ — UroMems, a global company developing innovative, mechatronics technology to treat stress urinary incontinence (SUI), announced today that it has successfully met the six-month primary endpoint for the first-ever female patient implanted with the UroActive™ System, the first smart automated artificial urinary sphincter (AUS) to treat SUI. This milestone indicates a new era for millions of women suffering from SUI, and signals an exciting transition for surgeons treating SUI not only in France, where the female patient was treated, but also across Europe and the U.S. It also follows closely on the heels of the successful results of the complete treatment cohort of the first-in-man clinical feasibility study. Results of this clinical study will contribute to the design and implementation of UroMems’ pivotal clinical trial in Europe and the U.S.
“On behalf of the medical team who implanted the UroActive System in the first female patient, Drs. Christophe Vaessen, Aurelien Beaugerie and I are over the moon to see our first patient has returned to living life fully after years of struggling with SUI,” said Professor Emmanuel Chartier-Kastler. “This promising therapy is a breakthrough technology in treating SUI in both women and men.”
The primary outcome measures include the successful device activation and the rate of explants and revisions at six months. The first female patient has not only met the study’s primary endpoints by remaining revision-free but is also experiencing restored social continence. Follow-up on secondary measures, including leak rate values, has been extremely positive.
“As the leading advocacy organization in the U.S. supporting patients and caregivers with urinary incontinence, we hear from patients weekly looking for information on the safety and efficacy of artificial sphincters. Unfortunately, we have little to report back and support their requests as no one in the U.S. actively promotes this option for women,” said Steven Gregg, Ph.D., executive director of the National Association for Continence. “These results of the first UroActive System implanted in a female represent a promising development to treat stress urinary incontinence in both women and men. We’re excited about this first-of-its-kind development and look forward to UroMems’ pivotal trial results.”
Pending those results, the potential for U.S. surgeons to offer this new option is now on the horizon; skilled surgeons performing robotic-assisted surgeries such as sacrocolpopexies and hysterectomies may soon be able to add implanting UroActive to their standard practices. UroActive is the first smart active implant that treats SUI, powered by a MyoElectroMechanical System (MEMS). This innovative system is placed around the urethra in men and the bladder neck in women, controlled based on the patient’s activity, without the need for manual adjustments, intending to provide patients with ease of use and a better quality of life than current options.
“We are elated to reach this critical achievement contributing to the demonstration of the feasibility of the UroActive System to successfully treat women suffering from debilitating SUI,” said Hamid Lamraoui, UroMems chief executive officer and co-founder. “The compelling results of this first-in-female implant show the high potential of our technology, bringing us one step closer to delivering on the massive unmet need for women and physicians desperately seeking a better SUI treatment option.”
SUI, or involuntary urinary leakage, affects an estimated 40 million Americans and 90 million Europeans, and occurs when the pressure in the bladder exceeds that of the muscle (the sphincter) around the urethra, caused by activities involving high intra-abdominal pressure, like coughing, laughing and exercising. SUI significantly impacts quality of life, as it can be debilitating, and often leads to depression, low self-esteem and social stigma.
UroMems aims to restore the quality of life, dignity and self-esteem of millions of men and women worldwide suffering from poorly treated chronic conditions by the commitment to change the perception that these disorders are inevitable as one grows older and is simply something to endure with no real solution. UroMems is revolutionizing the treatment of SUI with smart active implants, using the latest technological advances in the field of embedded systems and micro-technologies for the development of its groundbreaking solutions.
About UroActive
UroActive is an active implantable electronic artificial urinary sphincter that is being developed to compensate for sphincter insufficiency in patients, both men and women, with SUI. It is based on a unique bionic platform using embedded smart, digital and robotic systems which, based on data collected from a patient, create a treatment algorithm that is specific for each patient’s needs. The UroMems technology platform is protected by more than 120 patents and is designed to overcome the limitations of current solutions by optimizing safety and performance, patient experience and surgeon convenience. STeP participation does not imply product authorization. UroActive has not received marketing authorization from the FDA and is not available for sale in the United States or in the EU.
For more information, please visit www.uromems.com.
Media Contact:
Shelli Lissick
651-276-6922
eGenesis and OrganOx Announce Successful Use of a Genetically Engineered Porcine Liver with a Human Donor
eGenesis and OrganOx Announce Successful Use of a Genetically Engineered Porcine Liver with a Human Donor
72-hour proof-of-concept procedure at Penn Medicine marks longest perfusion using genetically
engineered porcine organ designed for liver support
Cambridge, Mass. (January 18, 2023) – eGenesis, a biotechnology company developing human-compatible organs and cells for the treatment of organ failure, and OrganOx, a medical device company with a focus on the therapeutic applications of isolated organ perfusion, today announced the successful completion of an extracorporeal perfusion of a brain-dead research donor using a genetically engineered porcine liver. The donor was the first to be enrolled in the ongoing PERFUSE-2 study.
The study, made possible through the generosity of a donor family seeking to help other families through the advancement of clinical research, was carried out in collaboration with the University of Pennsylvania Transplant Institute and Gift of Life Donor Program. The perfusion was performed using the eGenesis liver, EGEN-5784, connected to the OrganOx extracorporeal liver cross-circulation (ELC) device to enable circulation of the donor’s blood through the porcine liver. Stable blood flow, pressure, and pH were achieved throughout the procedure in addition to robust bile production. No evidence of rejection was observed. The perfusion was electively stopped per-protocol at 72 hours with the liver appearing healthy.
The PERFUSE-2 study is being conducted to evaluate the feasibility of using this liver perfusion system to support patients suffering from liver failure. Annually in the US, over 300,000 patients are admitted with various forms of liver failure, requiring treatment in the acute setting. The efficacy of existing liver support options is limited, and patients in liver failure face a high risk of mortality. For some patients, utilizing a human-compatible, genetically engineered porcine whole liver to support the function of a patient’s decompensated liver may provide time for the recovery of the patient’s native liver or time to obtain a liver transplant. eGenesis and OrganOx are co-developing this technology and anticipate submitting an Investigational New Drug (IND) Application to the U.S. Food and Drug Administration (FDA) in 2024 to initiate a first-in-human clinical study.
The genetically engineered porcine liver used in this study carried the same genetics as the porcine kidneys used in the landmark preclinical study recently published in Nature. These edits include (1) knock out of three genes involved in the synthesis of glycan antigens implicated in hyperacute rejection, (2) insertion of seven human transgenes involved in the regulation of several pathways that modulate rejection: inflammation, innate immunity, coagulation, and complement, and (3) inactivation of the endogenous retroviruses in the porcine genome.
“We wish to extend our deepest gratitude to the donor and their family for enabling this important medical achievement and for paving the way for future work toward a potential solution for the many patients in need of life-saving liver support,” said Michael Curtis, Ph.D., President and Chief Executive Officer of eGenesis. “In addition to being a first in the field of xenotransplantation, this study provides important information to help advance our IND application.”
Abraham Shaked, M.D., Ph.D. of the University of Pennsylvania Transplant Institute said, “The success of this study provides support for further exploration of organ products developed using advanced genome engineering to provide novel, high-quality therapeutic options for individuals experiencing organ failure.”
“This marks a key milestone in our journey towards an effective treatment for acute liver decompensation. The OrganOx ELC system combined with a genetically engineered liver from eGenesis links modern organ perfusion technology with the functions of a whole liver, with the goal of providing a treatment that offers a lifeline to the critically ill patient – time for their own liver to recover or time to receive a transplant,” said Prof Peter Friend, Chief Medical Officer, OrganOx.
“Gift of Life Donor Program, one of the nation’s leading organ procurement organizations, is proud to have collaborated on this pioneering study that has the potential to bring new hope to waitlist patients by providing a bridge to transplant or time for healing,” said Richard D. Hasz, Jr., MFS, CPTC, President and CEO, Gift of Life Donor Program. “This unique study was only possible thanks to the generosity of a donor family willing to help alleviate the suffering of others despite their own personal loss. Every day, we are inspired by the kindness of families who choose donation. We look forward to partnering with Penn Medicine, eGenesis and OrganOx on future trials to advance this evolving field of medicine.”
“Our family is very proud to support this medical advancement and see our loved one’s legacy benefit countless others,” said a member of the donor family. “It is a testament to our loved one’s selflessness and compassion to know this donation offers such hope for people suffering serious disease in the future.”
About OrganOx
OrganOx is a commercial stage UK-based medical device company with a focus on the therapeutic applications of isolated organ perfusion. Its first product, the OrganOx metra normothermic liver machine perfusion system, has been used to support more than 3000 liver transplant operations globally, optimizing the use of donated organs by enabling assessment of the quality of livers as well as longer preservation durations. Other therapeutic applications, including in kidney transplantation, are in development. Learn more at www.organox.com
About eGenesis
eGenesis is pioneering a genome engineering-based approach in the development of safe and effective transplantable organs. The eGenesis Genome Engineering and Production (EGEN™) Platform is the only technology of its kind to comprehensively address cross-species molecular incompatibilities and viral risk via genetic engineering. eGenesis has demonstrated durable preclinical success to date and is advancing development programs for acute liver failure, kidney transplant, and pediatric as well as adult heart transplant. Learn more at www.egenesisbio.com.
Media Contact
eGenesis Kimberly Ha
OrganOx
Daniel Woodward
First In Vivo CAR-M Lead Candidate Nominated Under Carisma-Moderna Collaboration
First In Vivo CAR-M Lead Candidate Nominated Under Carisma-Moderna Collaboration
First lead candidate to address a solid tumor indication with significant unmet medical need
Supportive pre-clinical proof of concept data reported at SITC 2023 that demonstrated feasibility, tolerability and early efficacy of in vivo CAR-M therapy utilizing mRNA/LNPs in solid tumors
PHILADELPHIA, Dec. 14, 2023 /PRNewswire/ — Carisma Therapeutics Inc. (Nasdaq: CARM) (“Carisma” or the “Company”), a clinical-stage biopharmaceutical company focused on discovering and developing innovative immunotherapies, today announced the nomination of its first lead candidate under the collaboration with Moderna, Inc. (Nasdaq: MRNA). This first lead candidate will target an antigen present on a solid tumor with significant unmet medical need. This strategic collaboration brings together Carisma’s chimeric antigen receptor macrophage (CAR-M) platform with Moderna’s messenger RNA (mRNA) and lipid nanoparticle (LNP) technologies to generate and develop in vivo CAR-M therapeutics for oncology.
“Following the compelling pre-clinical proof of concept data shared at SITC 2023, we believe in vivo CAR-M therapeutics that utilize Moderna’s mRNA/LNPs have the potential to benefit patients with a broad variety of cancers,” stated Michael Klichinsky, PharmD, PhD, Co-Founder and Chief Scientific Officer at Carisma. “The delivery of this first candidate demonstrates our ability to create novel in vivo CAR therapies that can be advanced toward the clinic. We are proud of the significant contributions made by both of the scientific teams at Carisma and Moderna towards this exciting program. We look forward to completing IND-enabling studies with the lead candidate and are excited about the prospect of bringing this therapy forward for patients with advanced solid tumors together with Moderna.”
Lin Guey, PhD, Chief Scientific Officer of Therapeutics Research Ventures and Biotherapeutics at Moderna, stated, “We are excited with the progress we’ve made to advance in vivo cell therapy (CAR-M) in collaboration with Carisma. Combining Carisma’s deep expertise in myeloid cell biology with our mRNA/LNP platform has allowed us to quickly advance the first lead candidate and we look forward to furthering its development, along with our continued collaboration with Carisma to develop novel therapies to treat patients.”
Pre-clinical proof of concept data were recently presented at SITC 2023, demonstrating the feasibility, efficacy, and tolerability of the in vivo CAR-M platform that utilizes Moderna’s optimized mRNA encapsulated in LNPs and is designed to redirect endogenous myeloid cells to exert targeted anti-tumor activity. The highlighted data demonstrated that Carisma’s CAR-M therapy can be directly produced in vivo, or within the body, and can successfully redirect endogenous myeloid cells against tumor-associated antigens using Moderna’ mRNA/LNPs. This novel approach to cancer immunotherapy offers an off-the-shelf solution that has the potential to increase access to CAR-based therapies and to be the basis of any CAR-M programs discovered or developed under the Carisma and Moderna collaboration.
About Carisma
Carisma Therapeutics Inc. is a clinical stage biopharmaceutical company focused on utilizing our proprietary macrophage and monocyte cell engineering platform to develop transformative immunotherapies to treat cancer and other serious diseases. We have created a comprehensive, differentiated proprietary cell therapy platform focused on engineered macrophages and monocytes, cells that play a crucial role in both the innate and adaptive immune response. Carisma is headquartered in Philadelphia, PA. For more information, please visit www.carismatx.com.
Cautionary Note on Forward-Looking Statements
Statements in this press release about future expectations, plans and prospects, as well as any other statements regarding matters that are not historical facts, may constitute “forward-looking statements” within the meaning of The Private Securities Litigation Reform Act of 1995. These statements include, but are not limited to, statements relating to Carisma’s business, strategy, future operations, cash runway, the advancement of Carisma’s product candidates and product pipeline, and pre-clinical and clinical development of Carisma’s product candidates, including expectations regarding timing of initiation and results of pre-clinical and clinical trials. The words “anticipate,” “believe,” “contemplate,” “continue,” “could,” “estimate,” “expect,” “goals,” “intend,” “may,” “might,” “outlook,” “plan,” “project,” “potential,” “predict,” “target,” “possible,” “will,” “would,” “could,” “should,” and similar expressions are intended to identify forward-looking statements, although not all forward-looking statements contain these identifying words.
Any forward-looking statements are based on management’s current expectations of future events and are subject to a number of risks and uncertainties that could cause actual results to differ materially and adversely from those set forth in, or implied by, such forward-looking statements. For a discussion of these risks and uncertainties, and other important factors, any of which could cause Carisma’s actual results to differ from those contained in the forward-looking statements, see the “Risk Factors” set forth in the Company’s Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on November 9, 2023, as well as discussions of potential risks, uncertainties, and other important factors in Carisma’s other recent filings with the Securities and Exchange Commission. Any forward-looking statements that are made in this press release speak as of the date of this press release. Carisma undertakes no obligation to revise the forward-looking statements or to update them to reflect events or circumstances occurring after the date of this press release, whether as a result of new information, future developments or otherwise, except as required by the federal securities laws.
Contacts
Investors:
Shveta Dighe
Head of Investor Relations
Media:
Julia Stern
(763) 350-5223
SOURCE Carisma Therapeutics Inc.
UroMems Reaches Significant Milestone: Successful Results in Clinical Feasibility Study of UroActive™ Smart Implant for Stress Urinary Incontinence Treatment
UroMems Reaches Significant Milestone: Successful Results in Clinical Feasibility Study of UroActive™ Smart Implant for Stress Urinary Incontinence Treatment
Successful primary and secondary results demonstrate proof of feasibility in male patients, paving way for launch of large-scale pivotal clinical study in the U.S. and Europe
GRENOBLE, France and MINNEAPOLIS, Dec. 13, 2023 /PRNewswire/ — UroMems, a global company developing innovative, mechatronics technology to treat stress urinary incontinence (SUI), announced today it has reached a significant milestone: the complete treatment cohort in the first-of-its-kind clinical feasibility study has successfully reached the six-month primary endpoints.
The feasibility assessment of the UroActive System was completed through a prospective multicenter clinical study. UroActive is the first smart automated artificial urinary sphincter (AUS) to treat SUI, and the only one to reach this critical milestone. The results of this initial clinical study support design and implementation of UroMems’ pivotal SUI trial in Europe and the U.S. All six men are now implanted for at least seven months and up to fifteen months, with their devices operating as expected and no need for revision nor explant. In addition, extremely positive follow-up was received on secondary outcomes measures, including leak rate values and patient quality of life questionnaires.
UroMems received very positive feedback from all patients participating in the study cohort. “You have changed my life,” stated one of the study participants. “I can do all activities again, without stress, without anxiety!”
“We’re so pleased to see that our expectations about our device’s performance were met or even exceeded and delighted to successfully treat and receive such high praise from patients who had been suffering from SUI for years,” said Professor Pierre Mozer, UroMems chief medical officer and co-founder. “The results of this study will allow us to prepare our pivotal study which will be a major step in the development of UroActive.”
UroActive is the first smart active implant that treats SUI, powered by a MyoElectroMechanical System (MEMS). This innovative system is placed around the urethral duct and is controlled based on the patient’s activity, without the need for manual adjustments, intending to provide patients with ease of use and a better quality of life than current options.
“The very compelling results of this first-in-man clinical study demonstrate the high potential of our technology and pave the way for larger clinical trials that will allow us to demonstrate all the benefits we are expecting to offer patients suffering from debilitating SUI,” said Hamid Lamraoui, UroMems chief executive officer and co-founder. “We could not have reached this important milestone without the enthusiastic participation of the men in this initial study cohort. We’re extremely grateful to them for being a vital part in bringing this potentially revolutionary treatment to market.”
SUI, or involuntary urinary leakage, affects an estimated 40 million Americans and 90 million Europeans, and occurs when the pressure in the bladder exceeds that of the muscle (the sphincter) around the urethra, caused by activities involving high intra-abdominal pressure, like coughing, laughing and exercising. SUI significantly impacts quality of life, as it can be debilitating, and often leads to depression, low self-esteem and social stigma.
About UroActive
UroActive is an active implantable electronic artificial urinary sphincter that is being developed to compensate for sphincter insufficiency in patients, both men and women, with SUI. It is based on a unique bionic platform using embedded smart, digital and robotic systems which, based on data collected from a patient, create a treatment algorithm that is specific for each patient’s needs. The UroMems technology platform is protected by more than 120 patents and is designed to overcome the limitations of current solutions by optimizing safety and performance, patient experience and surgeon convenience. STeP participation does not imply product authorization. UroActive has not received marketing authorization from the FDA and is not available for sale in the United States or in the EU.
For more information, please visit www.uromems.com.
Media Contact:
Shelli Lissick
651-276-6922
SOURCE UroMems

Nature Medicine Publication Highlights Potential for ONWARD® ARC Therapy™ to Improve Mobility After Parkinson’s Disease
Nature Medicine Publication Highlights Potential for ONWARD® ARC Therapy™ to Improve Mobility After Parkinson’s Disease
Therapy to be supported by newly awarded $1M grant from Michael J. Fox Foundation
EINDHOVEN, the Netherlands — November 06, 2023 — ONWARD Medical N.V. (Euronext: ONWD), the medical technology company creating innovative spinal cord stimulation therapies to restore movement, function, and independence in people with spinal cord injury (SCI), today announces a publication in Nature Medicine using investigational ONWARD ARC Therapy to address gait challenges related to Parkinson’s disease.
The study participant described in the Nature Medicine publication has been living with Parkinson’s disease for nearly three decades. He has a severe gait disorder that has not responded to conventional therapies.
“I could hardly walk without frequent falls, several times a day. In certain situations, like getting into an elevator, I would stomp, freeze as they say,” said the study participant in a press release issued by Switzerland’s Lausanne University Hospital (CHUV).
In 2021, researchers from the Swiss Federal Institute of Technology (EPFL) and CHUV investigated the possibility that ONWARD ARC Therapy (precise, targeted electrical stimulation of the spinal cord) could address common side effects of Parkinson’s disease that negatively impact mobility. The team collaborated with Dr. Erwan Bezard, a renowned neuroscientist from France’s National Institute of Health and Medical Research (INSERM).
After the introduction of ARC Therapy and benefitting from a few weeks of rehabilitation, the study participant was able to walk without previously noticeable gait interruptions. Today, he uses ARC Therapy eight hours per day.
“I turn on the stimulation in the morning and turn it off in the evening. It allows me to walk better, to stabilize myself. Even stairs don’t scare me anymore. Every Sunday I go to the lake, and I walk about six kilometers. It’s awesome,” said the participant in the CHUV release.
“The breakthrough reported in Nature Medicine shows the remarkable potential to use the same technology platform and therapy we are developing for spinal cord injury to also address mobilitychallenges stemming from Parkinson’s disease,” says Dave Marver, CEO of ONWARD.
“It is impressive to see that by electrically stimulating the spinal cord the same way we have done in paraplegic patients, we may be able to address gait disorders due to Parkinson’s disease,” adds neurosurgeon Jocelyne Bloch, co-director with Professor G. Courtine of ONWARD research partner .NeuroRestore.
.NeuroRestore was awarded a $1 million grant from The Michael J. Fox Foundation for Parkinson’s Research (MJFF) to implant the ARC-IM System and investigate the effect of ARC Therapy in six additional participants with Parkinson’s disease. This study will assist ONWARD in determining whether to conduct additional clinical trials and potentially commercialize ARC Therapy in the future for those living with Parkinson’s disease.
“MJFF is committed to fulfilling the unmet needs of people living with Parkinson’s disease by ensuring the development of improved therapies,” said Katharina Klapper, Director of Clinical Research, MJFF. “The Foundation is pleased to award a grant for scientists at EPFL and ONWARD Medical to investigate the effects of ARC Therapy in people with experiencing gait challenges.”
All ONWARD devices and therapies, including but not limited to ARC-IM, ARC-EX, and ARC Therapy, are investigational and not available for commercial use.
Note: ONWARD has previously disclosed achievement of human proof-of-concept for Parkinson’s disease in its Company Presentation.
About ONWARD Medical
ONWARD is a medical technology company creating therapies to restore movement, function, and independence in people with spinal cord injury (SCI) and movement disabilities. Building on more than a decade of science and preclinical research conducted at leading neuroscience laboratories, the Company has received nine Breakthrough Device Designations from the US Food and Drug Administration for its ARC Therapy™ platform.
ONWARD® ARC Therapy, which can be delivered by external ARC-EX™ or implantable ARCIM™ systems, is designed to deliver targeted, programmed spinal cord stimulation. Positive results were presented in 2023 from the Company’s pivotal study, called Up-LIFT, evaluating the ability for transcutaneous ARC Therapy to improve upper extremity strength and function. The Company is now preparing regulatory approval submissions for ARC-EX for the US and Europe. In parallel, the Company is conducting studies with its implantable ARC-IM platform, which demonstrated positive interim clinical outcomes for improved blood pressure regulation, a component of hemodynamic stability following SCI. Other ongoing studies include combination use of ARC-IM with a brain-computer interface (BCI).
Headquartered in Eindhoven, the Netherlands, ONWARD has a Science and Engineering Center in Lausanne, Switzerland and a US office in Boston, Massachusetts. The Company also has an academic partnership with .NeuroRestore, a collaboration between the Swiss Federal Institute of Technology (EPFL), and Lausanne University Hospital (CHUV).
For more information, visit ONWD.com, and connect with us on LinkedIn and YouTube.
For Media Enquiries:
Aditi Roy, VP Communications
For Investor Enquiries:
Khaled Bahi, CFO
Disclaimer
Certain statements, beliefs, and opinions in this press release are forward-looking, which reflect the Company’s or, as appropriate, the Company directors’ current expectations and projections about future events. By their nature, forward-looking statements involve several risks, uncertainties, and assumptions
that could cause actual results or events to differ materially from those expressed or implied by the forward-looking statements. These risks, uncertainties, and assumptions could adversely affect the outcome and financial effects of the plans and events described herein. A multitude of factors including, but not limited to, changes in demand, competition, and technology, can cause actual events, performance, or results to differ significantly from any anticipated development. Forward-looking statements contained in this press release regarding past trends or activities should not be taken as a representation that such trends or activities will continue in the future. As a result, the Company expressly disclaims any obligation or undertaking to release any update or revisions to any forward-looking statements in this press release as a result of any change in expectations or any change in events, conditions, assumptions, or circumstances on which these forward-looking statements are based. Neither the Company nor its advisers or representatives nor any of its subsidiary undertakings or any such person’s officers or employees guarantees that the assumptions underlying such forward-looking statements are free from errors nor does either accept any responsibility for the future accuracy of the forward-looking statements contained in this press release or the actual occurrence of the forecasted developments. You should not place undue reliance on forward-looking statements, which speak only as of the date of this press release. All ONWARD devices and therapies referenced here, including but not limited to ARC-IM, ARC-EX, and ARC Therapy, are investigational and not available for commercial use.
Carisma Presents Pre-Clinical Proof of Concept for in vivo CAR-M using mRNA platform in collaboration with Moderna at SITC
Carisma Presents Pre-Clinical Proof of Concept for in vivo CAR-M using mRNA platform in collaboration with Moderna at SITC
- Data demonstrate feasibility, tolerability, and early efficacy of mRNA/LNP in vivo CAR-M therapy in pre-clinical models of metastatic solid tumors
- Proof of concept achieved for platform under the Moderna collaboration
- "In vivo CAR-M: Redirecting endogenous myeloid cells with mRNA for cancer immunotherapy" to be presented Friday, November 3, 2023, at 11:30 am PT
PHILADELPHIA, Oct. 31, 2023 /PRNewswire/ — Carisma Therapeutics Inc. (Nasdaq: CARM) (“Carisma” or the “Company”), a clinical stage biopharmaceutical company focused on discovering and developing innovative immunotherapies, today announced that it will present new findings at the Society for Immunotherapy of Cancer (SITC) 38th Annual Meeting regarding its first-of-its-kind collaboration with Moderna. The collaboration aims to bring together Carisma’s chimeric antigen receptor macrophage (CAR-M) platform with Moderna’s mRNA and lipid nanoparticle (LNP) technologies to generate and develop in vivo CAR-M therapeutics.
Accepted as a late-breaking presentation, “In vivo CAR-M: Redirecting endogenous myeloid cells with mRNA for cancer immunotherapy,” showcases data that demonstrate Carisma’s CAR-M therapy can be directly produced in vivo, or within the body, successfully redirecting endogenous myeloid cells against tumor-associated antigens using mRNA/LNP. The pre-clinical data demonstrate feasibility, tolerability, and efficacy against metastatic solid tumors. This novel approach to cancer immunotherapy offers an off-the-shelf solution that has the potential to increase access to CAR-based therapies and will be the basis of CAR-M programs to be developed under the Carisma and Moderna collaboration.
“The data presented at SITC is incredibly exciting as it demonstrates that we have the ability to make CAR-M directly in vivo with mRNA/LNP technology, leading to robust and targeted anti-tumor activity,” said Michael Klichinsky, PharmD, PhD, Co-Founder and Chief Scientific Officer at Carisma. “This off-the-shelf approach to treat cancer with engineered myeloid cells, developed in collaboration with Moderna, has the potential to transform the CAR field and be applied to numerous cancer targets and indications.”
“We are pleased to share data about the successful application of our mRNA platform to advance in vivo cell therapy,” said Lin Guey, PhD, Chief Scientific Officer of External Research Ventures at Moderna. “We look forward to the continuation of our pre-clinical work with Carisma and are optimistic that the joint scientific knowledge of both companies will accelerate the development of novel in vivo CAR-M therapies for patients.”
Details of Carisma data accepted for presentation at the SITC 38th Annual Meeting are as follows:
- “In vivo CAR-M: Redirecting endogenous myeloid cells with mRNA for cancer immunotherapy” presented on Friday, November 3, 2023, at 11:30 am PT
- “CAR-Macrophages with custom intronic shRNA exhibit enhanced efficacy against solid tumors” presented on Friday, November 3, 2023, 9:00 am – 7:00 pm PT
- “Engineered Microenvironment Converters (EM-C): Macrophages expressing synthetic cytokine receptors reverse immunosuppressive signals in solid tumors” presented on Friday, November 3, 2023, 9:00 am – 7:00 pm PT
- “A Phase 1, First in Human (FIH) study of autologous macrophages engineered to express an anti-HER2 chimeric antigen receptor (CAR) in participants (pts) with HER2 overexpressing solid tumors” presented on Friday, November 3, 2023, 9:00 am – 7:00 pm PT
Presentation and posters will be available on the SITC 38th Annual Meeting portal for registered attendees.
About Carisma
Carisma Therapeutics Inc. is a clinical stage biopharmaceutical company focused on utilizing our proprietary macrophage and monocyte cell engineering platform to develop transformative immunotherapies to treat cancer and other serious diseases. We have created a comprehensive, differentiated proprietary cell therapy platform focused on engineered macrophages and monocytes, cells that play a crucial role in both the innate and adaptive immune response. Carisma is headquartered in Philadelphia, PA. For more information, please visit www.carismatx.com.
About Moderna
In over 10 years since its inception, Moderna has transformed from a research-stage company advancing programs in the field of messenger RNA (mRNA), to an enterprise with a diverse clinical portfolio of vaccines and therapeutics across seven modalities, a broad intellectual property portfolio and integrated manufacturing facilities that allow for rapid clinical and commercial production at scale. Moderna maintains alliances with a broad range of domestic and overseas government and commercial collaborators, which has allowed for the pursuit of both groundbreaking science and rapid scaling of manufacturing. Most recently, Moderna’s capabilities have come together to allow the authorized use and approval of one of the earliest and most effective vaccines against the COVID-19 pandemic.
Moderna’s mRNA platform builds on continuous advances in basic and applied mRNA science, delivery technology and manufacturing, and has allowed the development of therapeutics and vaccines for infectious diseases, immuno-oncology, rare diseases, cardiovascular diseases and auto-immune diseases. Moderna has been named a top biopharmaceutical employer by Science for the past eight years. To learn more, visit www.modernatx.com.
Cautionary Note on Forward-Looking Statements
Statements in this press release about future expectations, plans and prospects, as well as any other statements regarding matters that are not historical facts, may constitute “forward-looking statements” within the meaning of The Private Securities Litigation Reform Act of 1995. These statements include, but are not limited to, statements relating to Carisma’s business, strategy, future operations, cash runway, the advancement of Carisma’s product candidates and product pipeline, and pre-clinical and clinical development of Carisma’s product candidates, including expectations regarding timing of initiation and results of pre-clinical and clinical trials. The words “anticipate,” “believe,” “contemplate,” “continue,” “could,” “estimate,” “expect,” “goals,” “intend,” “may,” “might,” “outlook,” “plan,” “project,” “potential,” “predict,” “target,” “possible,” “will,” “would,” “could,” “should,” and similar expressions are intended to identify forward-looking statements, although not all forward-looking statements contain these identifying words.
Any forward-looking statements are based on management’s current expectations of future events and are subject to a number of risks and uncertainties that could cause actual results to differ materially and adversely from those set forth in, or implied by, such forward-looking statements. For a discussion of these risks and uncertainties, and other important factors, any of which could cause Carisma’s actual results to differ from those contained in the forward-looking statements, see the “Risk Factors” set forth in the Company’s Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on August 10, 2023, as well as discussions of potential risks, uncertainties, and other important factors in Carisma’s other recent filings with the Securities and Exchange Commission. Any forward-looking statements that are made in this press release speak as of the date of this press release. Carisma undertakes no obligation to revise the forward-looking statements or to update them to reflect events or circumstances occurring after the date of this press release, whether as a result of new information, future developments or otherwise, except as required by the federal securities laws.
Media Inquiries: